Cell lines and cell culture
HEK293T, U2OS, HeLa, IMR-90 and COS-7 cells were obtained from the University of California, Berkeley (UCB) cell culture facility and were grown in medium containing high-glucose DMEM (Thermo Fisher Scientific), 10% FBS (Sigma) and 1× penicillin–streptomycin (Thermo Fisher Scientific) at 37 °C with 5% CO2.
Plasmid construction
The nucleus-targeting Csm complex plasmid construction was described previously15 (Addgene, plasmid 195242). A cytoplasm-targeting Csm complex plasmid was generated from this by removing the NLS sequences before each protein sequence, changing dCsm–EGFP to dCsm–2×sfGFP and removing the U6-crRNA region. dPspCas13b–3×EGFP plasmid was purchased from Addgene (plasmid 132398) and dRfxCas13d–EGFP was modified from Addgene plasmid 109050 by removing the T2A sequence between dRfxCas13d and EGFP.
For the CRISPR array plasmid, NOTCH2 arrays were constructed through multiple steps of overlap extension PCR, as illustrated in Extended Data Fig. 2. Specifically, an oligo pool containing multiple 3–4-spacer fragments (sequences listed in Supplementary Table 1) was purchased from Integrated DNA Technologies (IDT). Then, the following steps were performed:
Step 1. Amplification of oligo pool fragments
A 50-µl PCR reaction was set up using Q5 high-fidelity 2× master mix (New England Biolabs (NEB)) containing 5 fmol of fragment (for example, spacers 1–4), 25 pmol of forward primer, 25 pmol of reverse primer, 25 µl of 2× master mix and water to a final volume of 50 µl. PCR was performed for 14–16 cycles. The PCR products were separated and purified from an agarose gel. If a smear was observed on the gel, the template amount or PCR cycle number was reduced.
Step 2. Joining of fragments
A 50-µl PCR reaction was set up using Q5 high-fidelity 2× master mix (NEB) containing 0.3 pmol of fragment 1 (for example, spacers 1–4 from step 1), 0.3 pmol of fragment 2 (for example, spacers 4–6 from step 1), 25 µl of 2× master mix and water to a final volume of 50 µl. Overlap extension PCR was performed for 6–8 cycles. The PCR products containing six spacers were separated and purified from an agarose gel.
Step 3. Addition of overhangs to joined fragments
A 50-µl PCR reaction was set up using Q5 high-fidelity 2× master mix (NEB) containing 5 fmol of joined fragment (for example, spacers 1–6 from step 2), 25 pmol of forward primer, 25 pmol of reverse primer (for example, containing overhang for spacer 7), 25 µl of 2× master mix and water to a final volume of 50 µl. PCR was performed for 14–16 cycles. The PCR products were separated and purified from an agarose gel.
Step 4. Joining of overhang-containing fragments
A 50-µl PCR reaction was set up using Q5 high-fidelity 2× master mix (NEB) containing 0.3 pmol of fragment 1 (for example, spacers 1–7 from step 3), 0.3 pmol of fragment 2 (for example, spacers 6–12 from step 3), 25 µl of 2× master mix and water to a final volume of 50 µl. Overlap extension PCR was performed for 6–8 cycles. The PCR products containing 12 spacers were separated and purified from an agarose gel.
Step 5. Cloning and sequence verification of intermediate fragments
The 12-spacer fragments from step 4 were cloned into vectors that were then introduced into bacteria for colony picking and sequence verification. Typically, 5–10 clones for each construct were sufficient to obtain a correct sequence.
Steps 6 and 7. Generation of final full-length arrays
Steps 3–5 were repeated to generate 24-spacer plasmids from the sequenced 12-spacer plasmids from step 5.
MAP1B arrays were constructed similarly but overhang regions were included in the original oligo pool sequences to bypass step 3 (Supplementary Table 1). Arrays were cloned downstream of a CAG promoter with a short signal sequence28 from the HSPB3 gene placed before the array to enhance pre-crRNA export from the nucleus. All cloning was performed in NEB stable Escherichia coli (NEB) to prevent recombination between repetitive sequences. Plasmids were verified by whole-plasmid sequencing. CrRNA and oligo pool sequences are listed in Supplementary Table 1. Plasmid sequences are listed in Supplementary Table 2.
Optical setup and image processing
Cell samples were imaged using a wide-field fluorescent microscope (Zeiss Axio Observer Z1 inverted fluorescence microscope) with a ×100/1.4 numerical aperture oil Ph3 Plan Apochromat objective, an ORCA-Flash4.0 camera (Hamamatsu), an X-Cite 120Q lamp and ZEN 2012 software. GFP filter sets included the BP 470/40 excitation filter, the FT 495 beamsplitter and the BP 525/50 emission filter. Atto590 and Alexa Fluor 568 filter sets included the BP 572/25 excitation filter, the FT 590 beamsplitter and the BP 629/62 emission filter. Images representing max-intensity z-projections were generated by FIJI software. Colocalization analysis was performed by FIJI plugin ComDet (version 0.5.5). Single-molecule tracking was performed by FIJI plugin TrackMate (version 7.12.1). The temporal color-coded images (Figs. 3e,f and 4b, e) were generated using the FIJI temporal color code function. Kymographs were generated using the KymoResliceWide plugin (version 0.6.0), with polyline selections used to track particle moving trajectories.
Immunostaining
For H2AK119ub staining, 1.5 × 105 HEK293T cells were grown on 18-mm-diameter, #1.5-thickness, collagen-coated coverslips (Neuvitro) in a 12-well plate. The next day, cells were transfected with 0.8 μg of XIST–targeting dCsm–GFP complex plasmid, 0.4 μg of dPspCas13b–3×EGFP plus 0.6 μg of XIST-targeting PspCas13b crRNA plasmid or 0.4 μg of dRfxCas13d–EGFP plus 0.6 μg XIST–targeting RfxCas13d crRNA plasmid using 5 μl of TransIT-293 transfection reagent (Mirus Bio). After transfection, cells were grown for 48 h to allow protein and crRNA expression. Then cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) in 1× PBS at room temperature for 10–15 min. Following three washes with 1× PBS, cells were permeabilized by 0.5% (v/v) Triton X-100 (Sigma) in 1× PBS for 10 min at room temperature. Samples were again washed with 1× PBS three times after permeabilization. The permeabilized cells were incubated in a blocking buffer (1× PBS containing 3% (w/v) BSA (Jackson ImmunoResearch)) for 1 h. Cells were then incubated with anti-H2AK119ub primary antibodies at 1:1,000 dilution (Cell Signaling, 8240S) in blocking buffer for 1 h at room temperature and washed with 1× PBS three times for 5 min each. Next, cells were stained with Alexa Fluor 568-labeled secondary antibodies in blocking buffer for 1 h at room temperature. Samples were washed again with 1× PBS three times to remove unbound antibodies. To prevent bound antibody dissociation, samples were postfixed with 4% (v/v) PFA in 1× PBS for 10 min and washed three times with 1× PBS for 5 min each.
For DCP1A staining, 1 × 105 U2OS cells were grown on 18-mm-diameter, #1.5-thickness, collagen-coated coverslips (Neuvitro) in a 12-well plate. The next day, 0.8 μg of cytoplasm-targeting Csm complex plasmid and two MAP1B CRISPR array plasmids (0.7 μg each) were transfected into cells using 5 μl of TransIT-LT1 transfection reagent (Mirus Bio). After transfection, cells were cultured for 48 h to allow protein and crRNA expression. Antibody staining was performed as for the above H2AK119ub procedure but with anti-DCP1A antibody (Abcam, ab183709).
RNA FISH
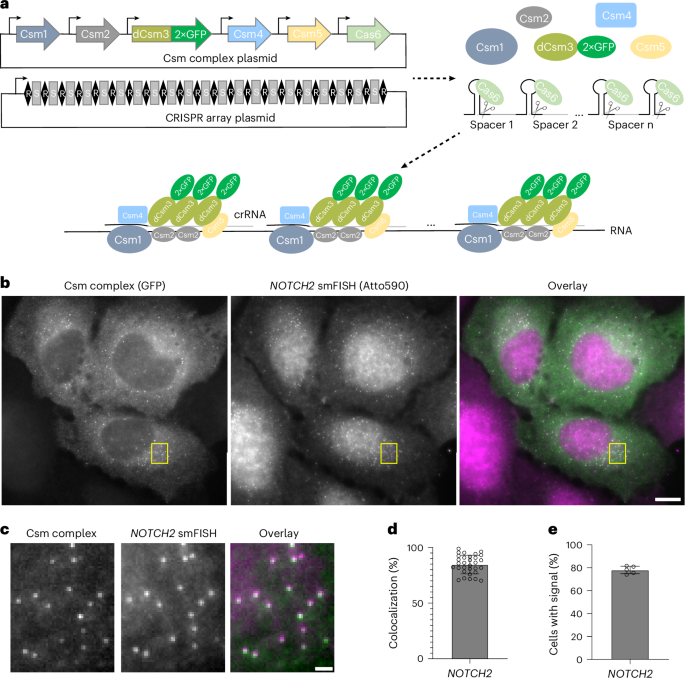
HEK293T, HeLa, IMR-90 and COS-7 cells were grown on 18-mm-diameter, #1.5-thickness, collagen-coated coverslips (Neuvitro) in a 12-well plate. The next day, 0.8 μg of cytoplasm-targeting Csm complex plasmid and two CRISPR array plasmids (0.7 μg each) were transfected into cells using 5 μl of TransIT-293 transfection reagent (Mirus Bio) or TransIT-LT1 transfection reagent (Mirus Bio). For U2OS cells, 1 × 106 cells were nucleofected with 1.5 μg of cytoplasm-targeting Csm complex plasmid and two CRISPR array plasmids (1.2 μg each). Then, U2OS cells were seeded on 18-mm-diameter, #1.5-thickness, collagen-coated coverslips (Neuvitro) at a density of 2.5 × 105 cells per well. After transfection, cells were grown for 48 h to allow protein and crRNA expression, fixed with 4% paraformaldehyde (Electron Microscopy Sciences) and permeabilized with 0.5% (v/v) Triton X-100 (Sigma) in 1× PBS for 10 min at room temperature. After a 5-min incubation in wash buffer comprising 2× SSC (Thermo Fisher Scientific) and 30% (v/v) formamide (Thermo Fisher Scientific), cells were stained with NOTCH2 or MAP1B mRNA FISH probes in hybridization buffer containing 30% (v/v) formamide, 0.1% (w/v) yeast tRNA (Thermo Fisher Scientific), 1% (v/v) murine RNase inhibitor (NEB), 10% (w/v) dextran sulfate (Sigma) and 2× SSC in a humidity-controlled 37 °C incubator overnight. FISH probes were applied at a concentration of 200 nM (5 nM per probe, ~40 probes in total). After staining, cells were washed twice with wash buffer at 37 °C, each for 30 min. Then, cells were stained with DAPI and 5 nM readout probes in a separate hybridization buffer composed of 2× SSC and 10% (v/v) ethylene carbonate (Sigma) in nuclease-free water before imaging. The NOTCH2 and MAP1B mRNA FISH probe sequences and readout probe sequence are provided in Supplementary Table 3. NOTCH2 and MAP1B FISH probes were ordered as oligo pools from IDT.
Live-cell imaging
For live-cell imaging of NOTCH2 and MAP1B mRNAs, 1 × 106 U2OS cells were nucleofected with 1.5 μg of cytoplasm-targeting Csm complex plasmid and two CRISPR array plasmids (1.2 μg each). Then, U2OS cells were seeded in a two-well glass-bottom NuncLab-Tek chamber (Thermo Fisher Scientific) at a density of 4 × 105 cells per well. After 48 h, the medium was changed to live-cell imaging buffer containing DMEM without phenol red supplied with 10% FBS, 1× penicillin–streptomycin and ProLong live antifade reagent (Thermo Fisher Scientific).
Puromycin treatment
For puromycin treatment, cells were incubated in live-cell imaging buffer containing 275 μM puromycin for 60 min at 37 °C before fixation or live-cell imaging.
Illustration software
Figures 2g and 3h were created using BioRender.com.
SMdM data analysis
SMdM analyses were described previously37. Briefly, single-molecule spots were first localized in all frames. Paired locations were identified across successive frames for calculation of displacements in the frame time Δt = 100 ms. The displacements were spatially binned with a grid size of 2.5 pixels (325 nm). The displacements in each spatial bin were separately fitted to a single-component diffusion mode through maximum likelihood estimation:
$$P(r)=\frac{2r}{a}\exp\left(-\frac{{r}^{2}}{a}\right)+br$$
(1)
Here, a = 4DΔt, where D is the diffusion coefficient and b accounts for a uniform background. The resultant local apparent D values were presented on a continuous color scale to produce a diffusivity map (Fig. 2c,d). Separately (Fig. 2e,f), all single-molecule displacements in each cell were pooled and fitted to a two-component diffusion mode50:
$$P(r)={F}_{1}\frac{2r}{{a}_{1}}\exp\left(-\frac{{r}^{2}}{{a}_{1}}\right)+(1-{F}_{1})\frac{2r}{{a}_{2}\,}\exp\left(-\frac{{r}^{2}}{{a}_{2}}\right)+br$$
(2)
where F1 and F2 = (1 − F1) are the fractions of the two diffusivity components and a1 = 4D1Δt and a2 = 4D2Δt account for the two diffusion coefficients D1 and D2.
RNA abundance measurements
Total cell RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific) as per the manufacturer’s instructions. Genomic DNA was removed using TURBO DNase (Thermo Fisher Scientific). After inactivating TURBO DNase with DNase-inactivating reagent, 2 μg of DNase-free RNA was reverse-transcribed using SuperScript III reverse transcriptase (Thermo Fisher Scientific) with random primers (Promega) as per the manufacturer’s instructions. qPCR was performed using iTaq Universal SYBR green supermix (Bio-Rad) in a CFX96 real-time PCR detection system (Bio-Rad). Gene-specific primer pairs used to detect mature transcripts are listed in Supplementary Table 4. The relative amount of target RNA compared to GAPDH was calculated using the 2−ΔΔCt method. Measurements were taken for three biological replicates, each with three technical replicates. No-RT and no-template controls were run alongside all RT–qPCR experiments.
RNA decay measurement
Cells were treated with 10 µg ml−1 actinomycin D (Thermo Fisher Scientific) for 0, 1, 2, 4, 8, 12 and 24 h to block transcription. Then, total RNA was extracted using the Direct-zol MiniPrep kit (Zymo Research) according to the manufacturer’s instructions. qPCR was performed on a CFX96 real-time PCR detection system (Bio-Rad) with the one-step RT–qPCR Kit (Thermo Fisher Scientific) to determine relative RNA levels. The relative mRNA levels of NOTCH2 and MAP1B versus the reference 18S ribosomal RNA were determined using three biological replicates. Following PCR amplification, melting curve analysis confirmed a single PCR product for each target gene. PCR primer sequences are listed in Supplementary Table 4.
Western blot
Cells were lysed in cold radioimmunoprecipitation assay lysis and extraction buffer (Thermo Fisher Scientific) supplemented with protease inhibitors (Sigma-Aldrich). Following centrifugation, the supernatants were collected and protein concentration measured using the Pierce 660-nm protein assay. Then, 10–30 µg of protein lysate was denatured in 1× Laemmli buffer at 95 °C for 10 min and resolved by SDS–PAGE. Proteins were transferred to an Immun-Blot LF PVDF membrane (Bio-Rad). The membrane was blocked with blocking buffer (0.05% Tween-20 and 3% BSA in 1× PBS) for 1 h at room temperature, incubated with primary antibody in blocking buffer for 2 h at room temperature, washed three times with 1× PBS, incubated with dye-conjugated secondary antibody for 1 h at room temperature and washed three more times with 1× PBS. The 700-nm and 800-nm channels of a LI-COR Odyssey CLx were used to visualize protein bands. The following primary antibodies were used for western blot: anti-NOTCH2 (Cell Signaling, 5732S; 1:1,000 dilution), anti-MAP1B (Thermo Fisher Scientific, PA5-82798; 1:1,000 dilution) and anti-ACTB (Proteintech, 60008-1-Ig; 1:2,500 dilution). The following secondary antibodies were used: IRDye 680RD goat anti-mouse (LI-COR, 926-68070; 1:20,000 dilution) and IRDye 800CW goat anti-rabbit (LI-COR, 926-32211; 1:20,000 dilution). Fiji was used to quantify the relative band intensities on blot images.
Statistics and reproducibility
Statistical analyses were conducted using GraphPad Prism (version 10.2.2). Exact statistical values are presented in the figures. The microscopy images presented from representative experiments were independently replicated at least three times with similar outcomes, unless explicitly indicated by the sample size noted in each figure.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.